
Актуальность исследования: Разработка методов клонирования и определения последовательности оснований (секвенирования) нуклеиновых кислот положила начало новому этапу развития молекулярной биологии. Знание первичной структуры участков генома, выполняющих определенные функции, дало возможность эффективно применить для их исследования целый арсенал новых методов генной инженерии. Эти методы (направленный мутагенез, рекомбинация in vitro и др.) позволяют модифицировать участки нуклеотидных последовательностей и исследовать их функции на молекулярном уровне. С их помощью комбинируются участки генетического материала и создаются геномы с совершенно новыми функциями [30].
К 1977 г. А. Максамом, У. Гилбертом и Ф. Сэнджером (Gilbert W., 1981; Sanger F., 1981) были разработаны специальные методы определения нуклеотидных последовательностей ДНК, которые получили название секвенирование (от англ. sequence — последовательность).
Эти методы сыграли судьбоносную роль в становлении геномики и генной инженерии. Методы секвенирования основаны на создании набора одноцепочечных фрагментов ДНК, оканчивающихся определенным нуклеотидом, для чего используются специфические рестриктазы. Разработаны разные методические подходы секвенирования и способы выделения набора фрагментов. В настоящее время высокий уровень технического оснащения сделал секвенирование достаточно рутинной лабораторной работой [12].
Сейчас многие исследователи продолжают разработку более емких и быстрых способов и подходов секвенирования и добиваются в этом значимых успехов. Таким образом, вполне закономерно, что в ближайшем будущем появятся еще более совершенные автоматические секвенаторы, что приведет к резкому увеличению числа расшифрованных последовательностей. Исходя из этого, изучение методов определения нуклеиновой последовательности ДНК становится особенно актуальным.
Благодаря знанию генетического кода появилась возможность определять участки нуклеотидных последовательностей, кодирующих потенциальные белки. Этот источник и сегодня дает нам основную информацию о функциональном строении нуклеотидной последовательности.
Цель работы: Изучить методы определения нуклеиновой последовательности ДНК.
Задачи работы:
- Дать определение нуклеиновой кислоты и изучить структуру дезоксирибонуклеиновой кислоты (ДНК);
- Рассмотреть метод секвинирования, его основные принципы и виды;
- Изучить методы Максама-Гилберта и Сегнера как основные методы секвенирования;
- Обозначить способы выявления достоверности секвенирования;
- Выделить новейшие методы определения последовательности ДНК.
12 стр., 5591 слов
Методология применения группы доходных методов при определении ...
... интеллектуальной собственности путем деления прибыли на ставку капитализации. 1.4 Метод освобождения от роялти Этот метод обычно используется для оценки стоимости патентов или лицензионных соглашений. ... между гипотетическим лицензиаром и лицензиатом. Основными этапами метода являются: 1. Определение размера прибыли, получаемой от использования интеллектуальной собственности. 2. Оценка возможного ...
1.
Нуклеиновые кислоты. Структура дезоксирибонуклеиновой кислоты (ДНК)
Прежде чем приступить к анализу методов секвенирования, необходимо определить понятие и значение нуклеиновых кислот, в частности ДНК.
Нуклеиновые кислоты — фосфорсодержащие биополимеры живых организмов, обеспечивающие хранение и передачу наследственной информации. Их макромолекулы состоят из неоднократно повторяющихся звеньев, которые представлены нуклеотидами. И их логично назвали полинуклеотидами [2].
Одной из главных характеристик нуклеиновых кислот является их нуклеотидный состав. В состав нуклеотида (структурного звена нуклеиновых кислот) входят три составные части:
Азотистое основание. Может быть пиримидиновое и пуриновое. В нуклеиновых кислотах содержатся основания 4-х разных видов: два из них относятся к классу пуринов и два — к классу пиримидинов.
Остаток фосфорной кислоты.
Моносахарид — рибоза или 2-дезоксирибоза. Сахар, входящий в состав нуклеотида, содержит пять углеродных атомов, т.е. представляет собой пентозу. В зависимости от вида пентозы, присутствующей в нуклеотиде, различают два вида нуклеиновых кислот — рибонуклеиновые кислоты (РНК), которые содержат рибозу, и дезоксирибонуклеиновые кислоты (ДНК), содержащие дизоксирибозу [3].
Нуклеотид по своей сути — это фосфорный эфир нуклеозида. В состав нуклеозида входят два компонента: моносахарид (рибоза или дезоксирибоза) и азотистое основание.
Азо́тистые основа́ния — гетероциклические органические соединения, производные пиримидина и пурина, входящие в состав нуклеиновых кислот. Для сокращенного обозначения пользуются большими латинскими буквами. К азотистым основаниям относят аденин (A), гуанин (G), цитозин (C), которые входят в состав как ДНК, так и РНК. Тимин (T) входит в состав только ДНК, а урацил (U) встречается только в РНК.В таблице 1 приведена структура главных азотистых оснований.
Таблица 1
Структура главных азотистых оснований
|
Азотистое основание |
|
|
|
|
|
|
Нуклеозид |
|
|
|
|
|
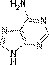 Аденин
Аденин 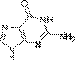 Гуанин
Гуанин 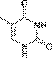 Тимин
Тимин 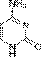 Цитозин
Цитозин 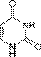 Урацил
Урацил 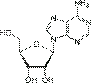 Аденозин A
Аденозин A 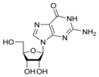 Гуанозин G
Гуанозин G 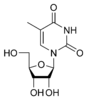 Тимидин T
Тимидин T 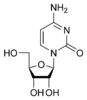 Цитидин C
Цитидин C 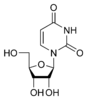 Уридин U
Уридин U Структура дезоксирибонуклеиновой кислоты (ДНК)
Первичная структура ДНК — порядок чередования дезоксирибонуклеозидмонофосфатов (дНМФ) в полинукпеотидной цепи.
Каждая фосфатная группа в полинукпеотидной цепи, за исключением фосфорного остатка на 5′-конце молекулы, участвует в образовании двух эфирных связей с участием 3′- и 5′-углеродных атомов двух соседних дезоксирибоз, поэтому связь между мономерами обозначают 3′, 5′-фосфодиэфирной [10].
Концевые нуклеотиды ДНК различают по структуре: на 5′-конце находится фосфатная группа, а на 3′-конце цепи — свободная ОН-группа. Эти концы называют 5′- и 3′-концами. Линейная последовательность дезоксирибонуклеотидов в полимерной цепи ДНК обычно сокращённо записывают с помощью однобуквенного кода, например -A-G-C-T-T-A-C-A- от 5′- к 3′-концу.
Вторичная структура ДНК. В 1953 г. Дж. Уотсоном и Ф. Криком была предложена модель пространственной структуры ДНК. Согласно этой модели, молекула ДНК имеет форму спирали, образованную двумя полинуклеотидными цепями, закрученными относительно друг друга и вокруг общей оси. Двойная спираль правозакрученная, полинуклеотидньхе цепи в ней антипараллельны, т.е. если одна из них ориентирована в направлении 3’→5′, то вторая — в направлении 5’→3′.
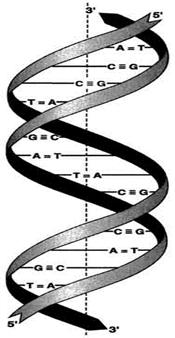
Двойная спираль ДНК. Молекулы ДНК состоят из двух антипараллельных цепей с комплементарной последовательностью нукпеотидов. Цепи закручены относительно друг друга в правозакрученную спираль так, что на один виток приходится примерно 10 пар нуклеотидов.
Молекулы ДНК расположены 5′-конец одной цепи и 3′-конец другой цепи [25].
Все основания цепей ДНК расположены внутри двойной спирали, а пентозофосфатный остов — снаружи. Полинуклеотидные цепи удерживаются относительно друг друга за счёт водородных связей между комплементарными пуриновыми и пиримидиновыми азотистыми основаниями А и Т (две связи) и между G и С (три связи).
При таком сочетании каждая пара содержит по три кольца, поэтому общий размер этих пар оснований одинаков по всей длине молекулы. Водородные связи при других сочетаниях оснований в паре возможны, но они значительно слабее. Последовательность нуклеотидов одной цепи полностью комплементарна последовательности нуклеотидов второй цепи. Комплементарые основания уложены в стопку в сердцевине спирали. Между основаниями двухцепочечной молекулы в стопке возникают гидрофобные взаимодействия, стабилизирующие двойную спираль.
Третичная структура ДНК (суперспирализация ДНК).
Каждая молекула ДНК упакована в отдельную хромосому. В диплоидных клетках человека содержится 46 хромосом. Общая длина ДНК всех хромосом клетки составляет 1,74 м, но она упакована в ядре, диаметр которого в миллионы раз меньше. Чтобы расположить ДНК в ядре клетки, должна быть сформирована очень компактная структура. Компактизация и суперспирализация ДНК осуществляются с помощью разнообразных белков, взаимодействующих с определёнными последовательностями в структуре ДНК. Все связывающиеся с ДНК эукариотов белки можно разделить на 2 группы: гисгоновые и негистоновые белки. Комплекс белков с ядерной ДНК клеток называют хроматином [19].
Методы исследования нуклеиновых кислот (молекулярно-генетические методы) — большая и разнообразная группа методов, в конечном счете, предназначенных для выявления вариаций в структуре исследуемого участка ДНК (аллеля, гена, региона хромосомы) вплоть до расшифровки первичной последовательности оснований [4].
В основе этих методов лежат «манипуляции» с ДНК и РНК. В результате бурного развития молекулярной генетики человека в 70-80-х годах и последующего успешного изучения генома человека молекулярно-генетические методы широко вошли в медико-генетическую практику. Часть ферментов, применяемых для исследования ДНК, представлена в таблице 2.
Основные ферменты, используемые в генной инженерии
|
Фермент |
Реакция |
Область приложениия |
|
Рестриктазы |
Расщепляют ДНК по специфическим последовательностям нуклеотидов |
Получение фрагментов ДНК, создание химерных молекул ДНК |
|
Нуклеаза |
Деградация как 5′-, так и 3 ‘-концов ДНК |
Образование концевых делеций в молекулах ДНК |
|
ДНК-лигаза |
Катализирует образование связей между молекулами ДНК |
«Сшивание» фрагментов ДНК |
|
ДНК-полимераза I |
Синтез двухцепочечной ДНК по ДНК-матрице |
Синтез двухцепочечной ДНК |
|
ДНКаза-I |
Вносит одноцепочечные разрывы в ДНК |
Картирование участков в ДНК |
|
Экзонуклеаза-III |
Удаляет нуклеотиды с 3 -концов ДНК |
Секвенирование ДНК |
|
Экзонуклеаза |
Удаляет нуклеотиды с 5-концов ДНК. |
Секвенирование ДНК |
|
Обратная транскриптаза |
Синтезирует ДНК по РНК-матрице |
Синтез кДНК по мРНК: картирование ДНК |
В последнее время молекулярная биология достигла больших высот, которые стали во многом возможны благодаря разработанным во второй половине 1970-х годов быстрым методам секвенирования ДНК. Слишком важной и нужной оказалась информация о последовательности нуклеотидов в ДНК. При этом следует отметить, что нынешнее секвенирование ДНК уже только напоминает то, что было в самом начале [10].
Исходя из стоящих перед конкретными исследователями задач, можно сказать, что есть разное секвенирование ДНК. Многие лаборатории проводят обычное секвенирование отдельных генов и прочих фрагментов ДНК, в то время как другие начинают проводить секвенирование полных геномов некоторых организмов. Такая процедура превратилась в настоящее производство, где многие операции полностью автоматизированы. Последнее является достижением уже не только молекулярной биологии, а и смежных областей науки и техники [6].
Так, секвенирование ДНК, включая многочисленные его составляющие, за четверть века своего эволюционирования претерпело целый ряд многочисленных преобразований и улучшений. На данном этаперазвития науки не существует единого мнения о том, как продуктивнее использовать методы секвенирования. Так, в обычном секвенировании ДНК на нынешнем этапе кто-то предпочитает использовать одноцепочечные матрицы ДНК, кто-то — двуцепочечные, другие же определяют последовательность ДНК с помощью только циклического секвенирования. Сходная ситуация наблюдается и с типом используемых меток, где выбор простирается от классического радионуклида 32Р до самых последних флуоресцентных красителей.
Итак, секвенирование — это общее название методов, которые позволяют установить последовательность нуклеотидов в молекуле ДНК. В настоящее время нет ни одного метода секвенирования, который бы работал для молекулы ДНК целиком; все они устроены так: сначала готовится большое число небольших участков ДНК (клонируется молекула ДНК многократно и «разрезается» её в случайных местах), а потом читается каждый участок по отдельности. Наработку интересующих последовательностей можно осуществить клонированием соответствующего фрагмента, например, методом ПЦР. Метод секвенирования по Максаму — Гилберту основан на химическом расщеплении ДНК по определенному основанию. Другой ферментативный метод (метод Сэнгера) базируется на применении аналогов нуклеотидов, прерывающих синтез комплементарной цепи ДНК по одноцепочечной матрице в месте встраивания в цепь соответствующего аналога. Секвенирование позволяет определить полную нуклеотидную последовательность всех хромосом, всего ДНК любого генома, любого организма. Этот метод позволяет определить последовательность нуклеотидов любых генов, что дает возможность их синтеза [21].
Клонирование происходит либо просто выращиванием клеток в чашке Петри, либо (в случаях, когда это было бы слишком медленно или по каким-то причинам не получилось бы) при помощи так называемой полимеразной цепной реакции. В кратком и неточном изложении работает она примерно так: сначала ДНК денатурируют, т.е. разрушают водородные связи, получая отдельные нити. Затем к ДНК присоединяют так называемые праймеры; это короткие участки ДНК, к которым может присоединиться ДНК-полимераза — соединение, которое, собственно, и занимается копированием (репликацией) нити ДНК. На следующем этапе полимераза копирует ДНК, после чего процесс можно повторять: после новой денатурации отдельных нитей будет уже вдвое больше, на третьем цикле — вчетверо, и так далее [22].
Все эти эффекты достигаются в основном с помощью изменений температуры смеси из ДНК, праймеров и полимеразы; для наших целей важно, что это достаточно точный процесс, и ошибки в нём редки, а на выходе получается большое число копий участков одной и той же ДНК. Разные методы секвенирования отличаются друг от друга не методами клонирования, а тем, как потом прочесть получившийся «суп» из многочисленных копий одной и той же ДНК.
Сейчас, все имеющиеся принципы и аналитические возможности современных методов секвенирования ДНК можно условно разделеть на три основных вида: классические — секвенирование с помощью капиллярного электрофореза и пиросеквенирование; новые («второго» поколения) — проводят одновременно секвенирование миллионов фрагментов ДНК, каждый из которых представлен кластером из многих тысяч или сотен тысяч своих клонов — это высокопроизводительное пиросеквенирование, циклическое лигазное и полупроводниковое секвенирование; секвенирование на молекулярных кластерах с использованием флуоресцентно меченных предшественников; новейшие («третьего» поколения) методы секвенирования, которые считывают информацию с миллионов единичных фрагментов ДНК без их предварительного клонирования. К ним относятся технология секвенирования одной молекулы, секвенирование единичных молекул в реальном времени и секвенирование через нанопоры. В нашем исследовании мы наиболее подобно рассмотрим два наиболее известных метода секвенирования: методы Максама-Гилберта и Сегнера. Также мы обсудим особенности новейших методов и технологий секвенирования [23].
3. Определение нуклеотидовой последовательности модифицированным методом Максама И Гилберта
Метод Максама и Гилберта основан на химической деградации ДНК. Он был предложен в 1976 году Максамом и Гилбертом и назван их именем. Суть метода сводится к следующему: один из концов фрагмента ДНК метят с помощью изотопа фосфора 32Р. В последнее время вместо радиоактивной вводят флюоресцирующую метку [1].
Ее можно «цеплять» и к нуклеотидам, причем для каждого типа нуклеотидов подбирать различную окраску. Препарат меченой ДНК делят на четыре порции и каждую из них обрабатывают реагентом, специфически разрушающим одно или два из четырех оснований, причем условия реакции подбирают таким образом, чтобы на каждую молекулу ДНК приходилось лишь несколько повреждений.
Разрушение идет в 2 этапа. На первом этапе происходит модификация азотистого основания и последующее выщепление его. На втором этапе производят гидролиз ДНК в местах выщепления оснований. Пуриновые основания модифицируются диметилсульфатом. Адениновые остатки метилируются по третьему атому азота, гуаниновые — по положению N7. Если такую модификацию обработать 0,1 М HCl при 0оС, то выщепляется метиладенин. При последующей инкубации в щелочной среде (0,1 М NaOH) при температуре +90оС происходит разрушение сахаро-фосфатной связи в местах выщепления оснований. Обработка поврежденных молекул пиперидином приводит к гидролизу ДНК по остаткам метилгуанина. Пиримидиновые основания модифицируются гидразином. В бессолевой среде модифицируется и цитозин, и тимин, в присутствии 2 М NaCl модифицируется только цитозин. При дальнейшей обработке пиперидином происходит расщепление ДНК по точкам модификации. Можно использовать и другие реакции химической модификации оснований и расщепления по ним молекул ДНК [31].
В результате получается набор меченых фрагментов, длины которых определяются расстоянием от разрушенного основания до конца молекулы. Фрагменты, образовавшиеся во всех четырех реакциях, подвергают электрофорезу в четырех соседних дорожках; затем проводят радиоавтографию, и те фрагменты, которые содержат радиоактивную метку, оставляют «отпечатки» на рентгеновской пленке. По положению отпечатков можно определить, на каком расстоянии от меченого конца находилось разрушенное основание, а зная это основание — его положение. Так набор полос на рентгеновской пленке определяет нуклеотидную последовательность ДНК. Аналогично наблюдают флюоресцентное окрашивание. Если для каждого из четырех нуклеотидов был подобран свой цвет флюоресцентной метки, то при электрофорезе их наносят на 1 дорожку. Тогда расположение нуклеотидов отмечено штрихами разного цвета, а процедуру считывания легко автоматизировать [1].
Реакции селективной модификации по каждому типу гетероциклических оснований проводятся таким образом, чтобы в каждой молекуле ДНК в среднем модифицировалось только одно звено данного типа. Поскольку все звенья данного типа в составе молекулы эквивалентны и реагируют с модифицирующим агентом с одинаковыми скоростями, то в сумме каждое звено этого типа окажется частично модифицированным. Дальнейшая обработка ДНК вторичным амином или щелочью приводит к отщеплению модифицированных гетероциклических оснований от цепи ДНК и разрыву полинуклеотидной цепи в местах отщепления гетероциклов (рис. 1) [2].
Модификации подвергают ДНК, 32Р-меченные по 5′-концевому нуклеотидному звену. Радиоактивная метка вводится фосфорилированием с помощью -32Р-АТР и Т4-полинуклеотидкиназы [5].
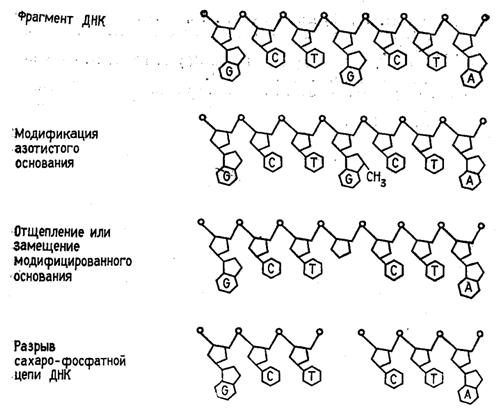
Рисунок 1. Отщепление модифицированных звеньев от цепи ДНК после обработки вторичным амином или щелочью.
Таким образом, в результате химической деградации получается набор фрагментов ДНК различной длины. Длины этих фрагментов соответствуют положению мономерных звеньев того типа, который подвергался модификации. Концевая радиоактивная метка служит точкой отсчета при определении длины продуктов химической деградации ДНК (рис. 2)
Набор полученных фрагментов фракционируется электрофорезом в ПААГ, который позволяет разделять олиго (поли) нуклеотиды, отличающиеся по длине всего на одно мономерное звено. Последовательность нуклеотидов в ДНК читается непосредственно с радиоавтографа геля [8].
Метод Максама и Гилберта, разработанный для анализа первичной структуры достаточно длинных ДНК, применим и для коротких (8 — 16 звенных) оли-годезоксирибонуклеотидов. Однако в этом случае реакции химической модификации проводят в более жестких условиях (увеличивая время и температуру реакции) с целью повышения степени модификации.
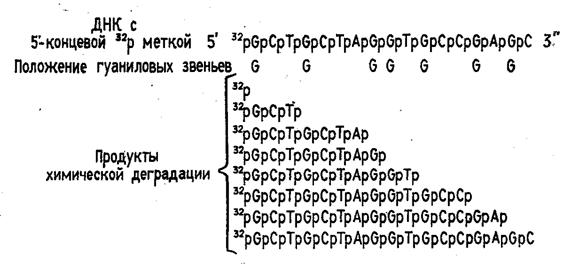
Рисунок 2. Химическая деградация ДНК.
Набор реакций, применяемых для расщепления ДНК по мономерным звеньям определенного типа достаточно велик и постоянно пополняется: по остаткам гуанина — обработка диметилсульфатом (рис. 3); по остаткам аденина и гуанина — апуринизация 50%-ной муравьиной кислотой (по Бартону); по остаткам аденина и цитозина — расщепление гетероциклических оснований под действием 1,2 н. гидроксида натрия и по остаткам тимидина и цитозина — обработка гидразином (рис. 4).
В настоящее время широко используются два основных варианта секвенирования по Максаму — Гилберту. В первом из них реакции химической модификации ДНК проводят в растворе, а во втором ДНК предварительно иммобилизуют на твердой фазе (например, ДЭАЭ-целлюлозе).
Первый метод более традиционен, его многочисленные модификации с успехом использовались для секвенирования фрагментов ДНК различных размеров, в том числе олигонуклеотидов. В то же время второй метод имеет ряд преимуществ. Он менее трудоемок и занимает меньше времени, проще в освоении, позволяет обойтись минимальным набором оборудования. В целом оба метода обеспечивают получение вполне приемлемых результатов, а выбор одного из них определяется конкретными условиями лаборатории.
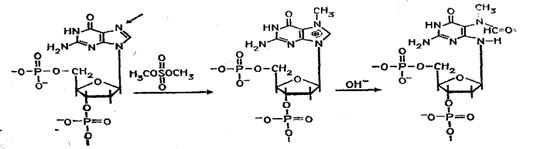
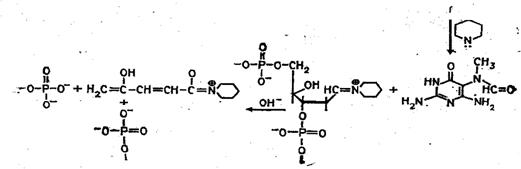
Рисунок 3. Реакция селективного расщепления по остаткам гуанина
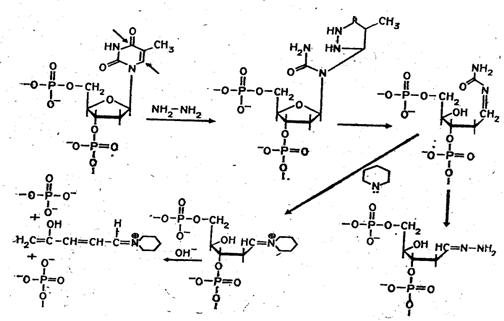
Рисунок 4. Реакция селективного расщепления по остаткам тимидина и цитозина.
Несмотря на относительно низкую производительность метода секвенирования ДНК путем химической деградации по Максаму-Гилберту в сравнении с ферментативным методом секвенирования ДНК по Сэнгеру, этот метод в настоящее время все же продолжает пользоваться популярностью. Так, метод химической деградации применяется для секвенирования синтетических олигонуклеотидов в тех случаях, когда это необходимо. Данный метод используют в том случае, когда особо «трудные» участки с сильной вторичной структурой не возможно секвенировать с помощью ферментативного построения новой цепи ДНК. К одному из преимуществ метода секвенирования ДНК химической деградацией можно отнести то, что здесь определяется последовательность фрагмента ДНК, или геномного, или клонированного, в каком-либо подходящем векторе (т.е. реплицировавшегося in vivo), а не новосинтезированная in vitro копия, как в ферментативном методе с дидезокситерминаторами. Еще одно отличие метода секвенирования ДНК по Максаму-Гилберту от метода Сэнгера <#»868987.files/image018.jpg»>
- Рисунок 5. Ферментативный метод секвенирования ДНК
Были также синтезированы модифицированные дидезоксирибонуклеотиды, в которых дезоксирибоза 3’-ОН отсутствует, для каждого из четырех оснований ДНК. ДНК-полимераза включает эти предшественники в ДНК. Однако, включившись в ДНК, модифицированное основание не может образовать фосфодиэфирную связь со следующим дезоксирибонуклеотидом. В результате рост (элонгация) данной цепи останавливается (терминируется) в том месте, где в ДНК включился дидезоксирибонуклеотид (ddNTP).
Поэтому их называют терминаторами элонгации
Реакционная смесь по Сэнгеру состоит из цепи ДНК, нуклеотидную последовательность которой надо определить, короткого фрагмента «меченой» ДНК, комплементарной концевому отрезку этой цепи (затравка), одного из четырех ddNTP и соответствующего dNTP в строго определенном соотношении (чтобы они конкурировали), а также остальных трех dNTP. Готовят четыре смеси, каждая из которых содержит один из четырех ddNTP. В каждой из пробирок образуется набор меченых фрагментов разной длины. Длина их зависит от того, в каком месте в цепь включен дефектный нуклеотид. Полученные меченые фрагменты ДНК разделяют в полиакриламидном геле (с точностью до одного нуклеотида), проводят радиоавтографию и по картине распределения фрагментов в четырех пробах устанавливают нуклеотидную последовательность ДНК [24].
Сразу вслед за разработкой быстрых методов секвенирования появились столь же быстрые и простые методы синтеза сравнительно длинных олигонуклеотидов с определенной, заранее заданной последовательностью. Теперь за три-четыре дня можно синтезировать последовательность из 12 — 20 нуклеотидов. Автоматизация этой процедуры еще более облегчает и ускоряет синтез. Появились приборы — ДНК-синтезаторы, которые выполняют эту работу за несколько часов.
В настоящее время выделение фрагментов ДНК, создание рекомбинантных генов, а так же прямое секвенирование ДНК и кДНК становятся общедоступными методами благодаря широкому внедрению ПЦР (полимеразной цепной реакции).
Сущность ПЦР заключается в использовании двух олигонуклеотидов-праймеров, способных специфически гибридизоваться с последовательностями нуклеотидов на противоположных концах двух цепей участка ДНК, в качестве затравки для одновременного синтеза комплементарных цепей с противоположных концов матрицы с помощью термостабильной ДНК-полимеразы. В ходе повторяющихся циклов (температурной денатурации ДНК, отжига и энзиматической достройки праймеров) экспоненциально увеличивается количество дискретного фрагмента, фланкированного последовательностями нуклеотидов, соответсвующих первичной структуре праймеров [23].
Применимость метода Сэнгера зависит от возможности получения одноцепочечных копий клонированных ДНК. Для этой цели можно использовать векторы на основе бактериофага М13. Двухцепочечную чужеродную ДНК можно клонировать в двухцепочечной репликативной форме (РФ) фаговой ДНК, при этом после трансформации в белковую оболочку будет упаковываться только одна из цепей ДНК. Во всех векторах типа М13тр используются сходные полилинкерные последовательности, поэтому для инициации полимеразных реакций пригоден один и тот же универсальный праймер. При амплификации смеси генов (например, семейства генов) необходимо провести клонирование ПЦР-продуктов в векторах типа М13, в результате каждый фаг будет содержать только одну вставку. При прямом секвенировании смеси генов наблюдается несколько одинаково расположенных полос в разных дорожках геля. При амплификации же одного гена можно проводить прямое секвенирование, не прибегая к промежуточному субклонированию[23] .
Выбор оптимального праймера для ПЦР зависит от 5 ‘- и 3 ‘-концевых последовательностей амплифицируемого фрагмента ДНК. Кроме того, для встраивания ПЦР-продукта в полилинкерный сайт вектора М13 в 5’-конец праймеров должны быть включены подходящие рестрикционные сайты. В этом случае ПЦР-амплификация с последующей рестрикцией продукта позволит провести его встраивание в ДНК М13, рестрицированную тем же ферментом. В разные концы амплифицируемого фрагмента лучше включать сайты для разных рестриктаз, поскольку это позволит избежать отжига векторной ДНК самой на себя и обеспечит положение клонированной вставки в определенной ориентации (так называемое направленное клонирование).
При подборе праймеров необходимо учитывать следующие факторы [27].
а. Следует убедиться в том, что амплифицируемое семейство генов не содержит консервативного внутреннего рестрикционного сайта, идентичного сайту, включенному в праймер.
б. После включения рестрикционного сайта 5′ — конец праймера нужно удлинить, в противном случае рестриктаза не будет расщеплять праймер. Необходимая для каждого фермента длина выступающего участка и время рестрикции указаны в каталоге фирмы New England BioLabs.
Перед секвенированием двухцепочечную рекомбинантную ДНК М13 необходимо перевести в одноцепочечную форму. Для этого ее вводят путем трансформации в компетентные клетки E. сoli. Бляшки, содержащие одноцепочечные рекомбинантные фаги, необходимо выколоть, нарастить в бактериальной культуре и депротеинизировать [27].
Затем переносят культуру в микроцентрифужную пробирку на 1,5 мл и центрифугируют в микроцентрифуге при 12 000 g в течение 5 минут. Переносят 1 мл супернатанта (содержащего чистый фаг) во вторую пробирку на 1,5 мл, добавляют 200 мкл полиэтиленгликоля и инкубируют при комнатной температуре как минимум 15 минут. Собирают фаг центрифугированием в течении 5 минут при 12 000 g и отбирают супернатант. Быстро повторяют центрифугирование и полностью удаляют все следы супернатанта. Затем осаждают ДНК ацетатом натрия, промывают ее 70%-ным этанолом и высушивают под вакуумом. Растворяют ДНК в 30 мкл воды. Полученная ДНК представляет собой одноцепочечную матрицу для секвенирования.
В настоящее время определение точной нуклеотидной последовательности любого сегмента ДНК умеренной длины — вполне разрешимая задача. Уже определена последовательность нескольких сотен генов про- и эукариот. Зная последовательность гена и генетический код, легко определить аминокислотную последовательность кодируемого им белка. Раньше для определения структуры белка приходилось делать тщательный и весьма трудоемкий анализ выделенного и очищенного белка. Сейчас часто бывает проще определить структуру белка через нуклеотидную последовательность, чем с помощью прямого секвенирования. Если секвенирование белка занимает месяцы и даже годы, то ДНК удается секвенировать за несколько недель.
Определение последовательности ДНК привело также к тому, что были обнаружены области, которые не кодируют белки, но принимают участие в регуляции экспрессии генов и репликации ДНК. В 1996 году был секвенирован геном дрожжей, в 1998 г. — геном арабидопсиса, в 2000 году — геном человека, однако в данном случае речь идет только об установлении последовательности нуклеотидов, так как генетическая структура и функции отдельных участков генома еще не идентифицированы, это более сложная задача [29].
Исходя из вышесказанного видно что, за время, прошедшее со дня разработки метода ферментативного секвенирования ДНК по Сэнгеру с помощью дидезокситерминаторов, он претерпел многочисленные модификации и всевозможные улучшения. В той или иной степени изменения подверглись практически все составляющие этого процесса. Так, другими стали подходы к получению матриц ДНК для их последующего секвенирования, практически не используется основной фермент первых лет — Кленовский фрагмент ДНК-полимеразы I E. Coli, изменились взгляды на затравочные молекулы, появились модифицированные дНТФ и ддНТФ, расширился выбор меченых молекул, включая и нерадиоактивные, заметно усовершенствовались этапы электрофоретического разделения, радиоавтографии и «чтения» нуклеотидной последовательности, несравненно выросло компьютерное обеспечение. Не говоря уже о появлении ПЦР и об автоматизации основных этапов определения нуклеотидной последовательности ДНК ферментативным методом, рассматриваемых, впрочем, в других главах [30].
В конечном итоге процесс секверирования по методу Сэнгера включает в себя: принцип секвенирования ДНК по Сэгнеру <http://enc.sci-lib.com/article0001401.html >, матрицы для секвенирования ДНК по Сэнгеру <http://enc.sci-lib.com/article0001402.html >, затравочные молекулы для ПЦР <http://enc.sci-lib.com/article0001407.html >, ДНК-полимераза <http://enc.sci-lib.com/article0001408.html >, мечение новосинтезируемой цепи ДНК <http://enc.sci-lib.com/article0001409.html > и терминирующие реакции по методу Сэнгера <http://enc.sci-lib.com/article0001410.html >.
Достоверность секвенирования ДНК
Немаловажное значение имеет достоверность получаемой информации в виде последовательности нуклеотидов, чему всегда уделялось серьезное внимание. Разработаны специальные компьютерные программы, направленные на выявление неточностей при анализе первичных данных, а также контролирующие весь ход выполнения проекта по крупномасштабному секвенированию ДНК. Особенно важным это становится в случаях когда в одной лаборатории выполняется более одного крупномасштабного проекта и поток данных чрезвычайно велик. Осуществлять контроль за такими экспериментами призвана программа Kaleidaseq [11].
На этапе сбора первичной информации процент ошибок сильно зависит от применяемого метода секвенирования, типа метки, используемого фермента, автоматического или ручного секвенатора и варьирует в зависимости от участка секвенируемой ДНК и опыта экспериментатора. В связи с точностью секвенирования особое значение приобретает и цель, стоящая перед исследователем. Так, при секвенировании коротких участков молекул кДНК, называемых ESTs и служащих идентификаторами генов, достоверности подобных данных серьезного значения не придается и, по некоторым оценкам, они могут содержать более 1% ошибок, что совершенно недопустимо в других случаях.
Первая группа была способна секвенировать ДНК с почти 100%-ной точностью в диапазоне 100-500 нуклеотидов, с 500 по 600 нуклеотид точность определения заметно снижалась. Для второй группы почти 100%-ная достоверность была доступна только в диапазоне 100-400, тогда как третья группа 92-100%-ную точность смогла обеспечить только в интервале 10Q-300 нуклеотидов. Однако на завершающем этапе точность секвенирования ДНК должна составлять, по крайней мере, 99,95%, что соответствует 5 ошибкам на 10000 нуклеотидов. Важно отметить, что в последнее время вполне достижимым стандартом считается 99,99% или 1 ошибка на 10000 нуклеотидов. Используемые в геномных проектах стратегии случайного подхода, и достигаемое за счет этого в среднем 7-8-кратное покрытие генома, кроме избыточных данных, дают существенное преимущество в виде многократного «чтения» одних и тех же участков (причем, по обеим цепям) ДНК, что приводит к необходимой достоверности получаемых результатов [32].
Современными исследователями была разработана программа, способная благодаря специальному алгоритму выявлять и исправлять в кодирующих регионах определенный тип ошибок, получивших общее обозначение «индел», в виде инсерций и делеций нуклеотидов, нарушающих рамку считывания. Недавно созданная программа PHRED позволяет осуществлять предсказание специфических «ошибок» в «прочтении» определенных нуклеотидов на этапе сбора первичной информации, что делает ее весьма удобной при выполнении крупномасштабных проектов. Так, с помощью данной программы был проведен анализ первичных данных 6 американских лабораторий, выполняющих проекты по секвенированию геномов. В результате оказалось, что предсказанные данной программой «ошибки» в определении тех или иных нуклеотидов во многом совпали с действительными, хотя справедливости ради следует отметить, что последних было все же несколько меньше. Ранее похожая программа, направленная на предсказание потенциальных ошибок в первичном материале секвенирования ДНК, была предложена другими авторами.
Таким образом, видно, что вопрос о достоверности секвенирования ДНК остается открытым. Не смотря на то, что исследователи достигли в этом направлении значительных успехов, сейчас продолжается разработка более точных программ достоверности.
6. Новейшие методы определения последовательности ДНК
Методы секвенирования третьего поколения (NNGS) призваны исправить основные недостатки прежних методов, а именно: сложную пробоподготовку, небольшую длину единичных прочтений, потребность в усилении сигнала от каждого из анализируемых фрагментов ДНК путем их амплификации, длительное время цикла, необходимость многочисленного повторного секвенирования.
Технология секвенирования одной молекулы.
В 2008 г. компания «Helicos BioSciences» впервые продемонстрировала на своем приборе метод секвенирования нуклеотидной последовательности единичных фрагментов ДНК без их предварительной амплификации [12].
Эта технология — «tSMS» (true Single Molecule Sequencing) обладает очень высокой чувствительностью, благодаря разработанному компанией сверхнизкому фоновому свечению поверхности подложки, реагентам для секвенирования и способу визуализации. Согласно новому методу, ДНК разделяется на небольшие фрагменты, и к одному из концов каждого фрагмента с помощью специального фермента прикрепляют длинную последовательность из молекул аденина, имеющую в конце светящуюся метку. На специальную подложку прикрепляют последовательности из молекул тимина на расстоянии, позволяющем анализировать каждый фрагмент по отдельности. Когда образцы распределяют на подложку, то происходит комплементарное связывание последовательностей из тимина, прикрепленных на ее поверхности, с адениновыми последовательностями, связанными с фрагментами ДНК. Далее подложка сканируется, и по свечению меток находят положение каждого присоединенного отрезка молекулы ДНК. Затем метку удаляют и добавляют по очереди каждый из четырех типов меченых нуклеотидов, детектируя свечение в местах связывания, после чего метки от присоединенных нуклеотидов удаляют и вводят следующий тип меченых нуклеотидов. Так, нуклеотид за нуклеотидом последовательность достраивается по комплементарной цепи ДНК, что позволяет секвенировать фрагменты длиной до 55 п.н.
Компьютер записывает положение миллионов вспышек после каждой реакции. Такая система обеспечивает прочтение миллиардов нуклеотидов в сутки с точностью до 99,999 % при 20-кратном перекрывании [12].
Однако прибор, основанный на данной технологии («Heli Scope»), получился слишком дорогим, расход реагентов высоким при малой длине чтения нуклеотидных последовательностей. В ноябре 2012 г. компания «Helicos Biosience» была признана банкротом и прекратила свое существование.
Секвенирование единичных молекул в реальном времени.
В 2009 г. был представлен метод секвенирования «Single Molecule RealTime» (SMRT), разработанный компанией «Pacific Biosciences». Секвенатор этой компании позволяет «читать» каждый фрагмент ДНК десятки раз, при этом консенсусная точность определения его нуклеотидной последовательности достигает 99,9 %. Технология данного метода основана на секвенировании в реальном времени однонитевых фрагментов ДНК длиной до 10000 п.н. и более с помощью ДНК-полимеразы [31].
На дно специальных ячеек, расположенных на прозрачном стекле, прикрепляются одиночные молекулы ДНК-полимеразы. Область вокруг закрепленного фермен-та просвечивается с помощью специального лазера.
В каждую ячейку добавляют нуклеотиды всех четырех типов, помеченные разными светящимися маркерами. Область, которую анализирует лазер, столь мала, что меченые нуклеотиды не задерживаются в ней достаточно долго, чтобы их свечение было зафиксировано прибором. Если же ДНК-фрагмент удерживается полимеразой, то во время достройки его комплементарной цепи сигнал от каждого присоединившегося нуклеотида фиксируется в сканируемой области. После присоединения очередного нуклеотида его светящаяся метка удаляется.
Прибор «PacBio RS», основанный на этой технологии, имеет высокую собственную стоимость, но в то же время экспрессную пробоподготовку, высокую скорость и низкую стоимость секвенирования ДНК .
Секвенирование через нанопоры.
Возможность использовать матрицы с нанопорами для быстрого секвенирования ДНК исследуется уже более 15 лет в научных центрах Европы и США. Нанопоры представляют собой наноотверстия, которые могут быть биологическими, например порообразующий белок в мембране как липидный бислой, или твердотельными (из таких синтетических материалов, как нитрид кремния или графен).
При секвенировании через нанопоры используется физическое различие между нуклеотидными основаниями для их идентификации в молекуле ДНК. Отрицательно заряженный одноцепочечный фрагмент ДНК длиной в несколько тысяч нуклеотидов протягивают через пору в мембране диаметром 2-5 нм, регистрируя изменение электропроводности на но поры при помощи электродов по мере поочередного прохождения через нее нуклеотидов. Каждому типу основания соответствует свое изменение электропроводности из-за различия между ними по размерам, поэтому они закрывают пору в большей или меньшей степени и на разную продолжительность. Соответственно этому изменяется и электропроводность. Однако данная технология имеет, по крайней мере, два серьезных технических препятствия для реализации: отсутствие надежного подхода к контролю продвижения ДНК через нанопоры и технические трудности в создании достаточно малых датчиков [12].
В одном из вариантов для замедления прохождения фрагмента ДНК через нанопоры на его конец крепят магнитную микросферу. Чтобы тянуть молекулу за микросферу, используют магнит, включенное электрическое поле в то же время тянет ДНК в противоположную сторону, затягивая ее в отверстие нанопоры. Таким образом, скорость движения ДНК через нанопоры определяется балансом этих сил.
При этом чтение последовательности нуклеотидов происходит со скоростью в сотни тысяч раз быстрее, по сравнению со стандартными методами секвенирования. Секвенирование генома человека займет в этом случае всего 20 ч, поскольку многократной амплификации матрицы не понадобится [11].
Другой предложенный фирмой «IBM» вариант технологии — «ДНК Транзистор», использует многослойную (диэлектрик-металл) мембрану. Изменение напряжения между адресными слоями металла в мембране создает электрические поля внутри нано-пор, циклически включая и выключая которые мож-но двигать ДНК через нанопоры с шагом в один ну-клеотид за цикл, улавливая датчиками разницу между возможными четырьмя вариантами оснований.
Существуют также другие варианты реализации секвенирования ДНК через нанопоры, но в настоящее время научно-исследовательские разработки нанопорных секвенаторов находятся на опытно-конструкторской стадии.
Недавно компания «Oxford Nanopore Technologies» заявила о том, что практически доработала технологию секвенирования на основе нанопор до ее коммерческого аппаратного воплощения. Выход первых нанопорных секвенаторов данной компании под названием «GridION» и «MinION» может состо-яться уже в 2015 г. Уровень ошибок данной технологии составит 0,1-1 %, а длина читаемых единичных фрагментов ДНК достигнет многих десятков тысяч нуклеотидов. Это будет очередным прорывом в области секвенирования ДНК, так как время, затрачиваемое на данный процесс, и его стоимость уменьшатся на порядок.
На сегодняшний день из практически реализованных методов секвенирования третьего поколения функционирует только платформа «SMRT» (Pacific Biosciences).
Вероятно, через несколько лет метод секвенирования ДНК через нанопоры станет доминирующим и вытеснит практически используемые методы второго поколения [12].
Таким образом, имеющиеся методы секвенирования направлены на выполнение разноплановых задач, связанных с определением нуклеотидной последовательности ДНК. Новейшие методы секвенирования могут в ближайшее время заменить своих предшественников, в том числе стать практической альтернативой и для классических методов секвенирования ДНК.
дезоксирибонуклеиновый кислота секвенирование последовательность
Секвенирование нуклеиновых кислот в настоящее время стало рутинным методом молекулярной биологии. Благодаря знанию генетического кода появилась возможность определять участки нуклеотидных последовательностей, кодирующих потенциальные белки. Этот источник дает основную информацию о функциональном строении нуклеотидной последовательности. Секвенирование — это общее название методов, принципов и технологий, которые позволяют определить полную нуклеотидную последовательность всех хромосом, всего ДНК любого генома, любого организма. Этот метод позволяет определить последовательность нуклеотидов любых генов, что дает возможность их синтеза.
Все имеющиеся принципы и аналитические возможности современных методов секвенирования ДНК имеют условную классификацию. Их можно разделить на три основных вида: классические — секвенирование с помощью капиллярного электрофореза и пиросеквенирование; новые —высокопроизводительное пиросеквенирование, циклическое лигазное и полупроводниковое секвенирование; секвенирование на молекулярных кластерах с использованием флуоресцентно меченных предшественников; новейшие − технология секвенирования одной молекулы, секвенирование единичных молекул в реальном времени и секвенирование через нанопоры. В нашем исследовании мы наиболее подобно рассмотрены два наиболее известных метода секвенирования: методы Максама-Гилберта и Сегнера. Также мы обсудим особенности новейших методов и технологий секвенирования.
Метод Максама-Гилберта и метод Сэнгера, основаны на одном принципе. В первом используется специфическое расщепление ДНК, обусловленное природой оснований, во втором — статистический синтез ДНК, заканчивающийся на каком-либо одном из 4 нуклеотидов. Таким образом, основой обоих методов является получение полного (статистического) набора фрагментов ДНК, оканчивающихся на каждом из четырёх нуклеотидов.
Химический метод (метод Максама-Гилберта) проще использовать в том случае, когда исследуемая ДНК не слишком велика (200-500 звеньев).
В том случае, если речь идет о секвенировании высокомолекулярной ДНК, лучше применять метод полимеразного копирования (метод Сэнгера) , чтобы не вводить процедуру рестриктазного расщепления с выделением индивидуальных фрагментов. При энзиматическом секвенировании протяженных одноцепочечных ДНК (например, бактериофагов) можно применять набор олигонуклеотидов-затравок, синтез которых в настоящее время не требует больших затрат времени и труда. Для двутяжевых высокополимерных ДНК наиболее удобен метод слепого энзиматического секвенирования с применением универсальной затравки (их выпускают многие фирмы) и обработки данных с помощью ЭВМ. Химический метод также может быть применен, но в этом случае необходимо вырезать из вектора исследуемые фрагменты ДНК, и это усложняет всю процедуру.
Относительно новейших методов секвенирования необходимо отметить, что они призваны исправить основные недостатки прежних методов, а именно: сложную пробоподготовку, небольшую длину единичных прочтений, потребность в усилении сигнала от каждого из анализируемых фрагментов ДНК путем их амплификации, длительное время цикла, необходимость многочисленного повторного секвенирования.
Таким образом, имеющиеся методы секвенирования направлены на выполнение разноплановых задач, связанных с определением нуклеотидной последовательности ДНК. Новейшие методы секвенирования могут в ближайшее время заменить своих предшественников, в том числе стать практической альтернативой и для классических методов секвенирования ДНК.
1. Александров А.А., Александров Н.Н., Бородовский М.Ю. Компьютерный анализ генетических текстов / А.А.Александров, Н.Н.Александров, М.Ю.Бородовский и др. − М., Наука, 2003.
2. Артюхов И.В., Кеменов В.Н., Нестеров С.Б. Биомедицинские технологии. Обзор состояния и направления работы. Материалы 9-й научно-технической конференции «Вакуумная наука и техника» / И.В. Артюхов, В.Н. Кеменов, С.Б. Нестеров. − М., МИЭМ, 2002.
3. Барановов В. С. Генная терапия — медицина XXI века // Соросовский образовательный журнал / В. С. Барановов. — М., 1999. − № 3.
- Бекер М. Е., Лиепиньш Г.К., Райпулис Е.П. Биотехнология / М. Е. Бекер, Г.К. Лиепиньш, Е.П. Райпулис. − М., Агропромиздат, 1990.
- Борисюк Н.В.
Молекулярно — генетическая конституция соматических гибридов // Биотехнология. Итоги науки и техники ВИНИТИ АН СССР / Н.В. Борисюк. − М., 1988. − Т. 9.
- Гауптман З., Грефе Ю., Ремане Х. Органическая химия / З. Гауптман, Ю. Грефе, Х. Ремане. — М., 2000.
- Глебов О.
К. Генетическая трансформация соматических клеток // Методы культивирования клеток / О. К. Глебов. — СПБ., Наука, 1988.
- Грин Н., Стаут У., Тейлор Д. Биология / Н. Грин, У. Стаут, Д. Тейлор. — СПб, 2000.
- Дейвис К.
Анализ генома. Методы / Под ред. К. Дейвиса. — М., Мир, 1990.
10. Жарких А.А. Методы филогенетического анализа генов и белков //Молек.биология. Итоги науки и техники. ВИНИТИ / А.А. Жарких. − М., 1985.
11. Зубов В.В. Секвенирование по Ротбергу (потенциал полупроводникового секвенирования) / В.В. Зубов. − СПБ., Биомика, 2013. − Том 5. − №1.
12. Краснов Я.М., Гусева Н.П., Шарапова Н.А., Черкасов А.В. Современные методы Секвенирования днк (обзор) // Проблемы особо опасных инфекций, вып. 2 / Я.М.Краснов, Н.П.Гусева, Н.А.Шарапова, А.В.Черкасов. − Саратов, ФКУЗ «Российский научно-исследовательский противочумный институт «Микроб, 2014.
- Курчанов Н.А. Генетика человека с основами общей генетики. Учебное пособие / Н.А. Курчанов. — СПб., СпецЛит, 2009.
14. Лещинская И. Б. Генетическая инженерия // Соросовский образовательный журнал / И. Б. Лещинская. — М., 1996. − №1.
15. Ли А., Тинланд Б. Интеграция т-ДНК в геном растений: прототип и реальность // Физиология растений / А. Ли, Б. Тинланд. — СПб., 2000. − том 47.− № 3.
- Микельсон А. Химия нуклеозидов и нуклеотидов / А. Микельсон. — М., 2000.
- Нестеров.
C.Б. Нанотехнология. Современное состояние и перспективы. «Новые информационные техноло-гии». Тезисы докладов XII Международной студенческой школы-семинара / C.Б. Нестеров. − М., МГИЭМ, 2004.
- Пехов А.П. Биология и общая гинетика /А.П. Пехов. — М., 2001.
- Пирузян Э.
С. Генетическая инженерия растений / Э.С. Пирузян. − М., Знание, 1988.
- Пирузян Э. С. Основы генетической инженерии растений / Э.С. Пирузян. − М., Знание, 1989.
- Попов Л.
С., Языков А. А. Трансгенные животные как модели для изучения репродукции эмбрионального развития и заболеваний человека // Успехи современной биологии / Л.С. Попов., А.А. Языков. — М., 1999. − Т 119. − № 1.
22. Прокофьев М.А. Экспериментальные методы исследования белков и нуклеиновых кислот / Под ред. М.А.Прокофьева. — М., Изд-во МГУ, 1985.
23. Разумовская И.В. Нанотехнология: Учеб. Пособие. Элективный Курс / И.В. Разумвская. − М., Дрофа, 2009.
24. Романов Г. А. Генетическая инженерия растении и пути решения проблемы биобезопасности // Физиология растений / Г. А. Романов. — М., 2000.
25. Салганник Р.И. Методы молекулярной генетики и генной инженерии. / Р.И. Салганник. — Новосибирск, Наука. Сиб. отд-ние, 1990.
26. Шабарова З.А., Богданов А.А. Химия нуклеиновых кислот и их компанентов / З.А. Шабарова, А.А. Богданов. — М., Химия, 1978.
27. Шабарова З.А., Богданов А.А. Химия нуклеиновых кислот и их полимеров / З.А. Шабарова, А.А. Богданов. — М., 2000.
28. Шабарова З.А., Богданов А.А., Золотухин А.С. Химические основы генной инженерии: Учебное пособие / З.А. Шабарова, А.А. Богданов. — М., Изд-во МГУ, 1994.
29. Щелкунов С. Н. Генетическая инженерия / С.Н. Щелкунов. — Новосибирск, Изд-во Новосибирского ун-та, 1994.
30. <http://allrefs.net/c12/3te0w/p18/?ful> l
- <http://www.bioconsulting.ru/e/3128740-novyiy-metod-opredeleniya-posledovatelnosti>
- <http://www.biotechnolog.ru/ge/ge6_1.htm>