
Московская Медицинская академия им. И.М.Сеченова
Кафедра медицинской генетики
Фенилкетонурия
лечебного факультета
33 группы Алиева С.М.
Москва
2004
ФЕНИЛКЕТОНУРИЯ — тяжелое наследственное заболевание, которое характеризуется главным образом поражением нервной системы.
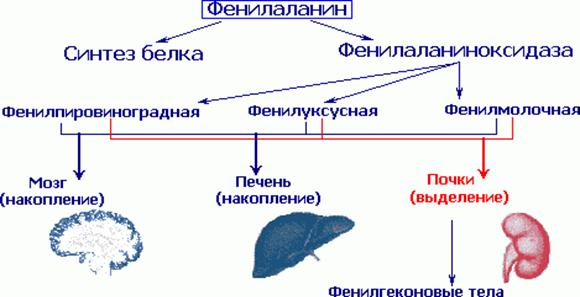
Этиология и патогенез. В результате мутации гена, контролирующего синтез фенилаланингидроксилазы, развивается метаболический блок на этапе превращения фенилаланина в тирозин, вследствие чего основным путем преобразования фенилаланина становится дезаминирование и синтез токсических производных — фенилпировиноградной, фенил-молочной и фенилуксусной кислот. В крови и тканях значительно увеличивается содержание фвнилаланина (до 0,2 г/л и более при норме 0,01-0,02 г/л).
Существенную роль в патогенезе болезни играет недостаточный синтез тирозина, который является предшественником катехоламинов и меланина. Заболевание наследуется по аутосомно-рецессивному типу.
Фенилкетонурия (ФКУ) — тяжелое наследственное заболевание, наступающее вследствие врожденного дефекта фермента, отвечающего в организме человека за нормальный обмен фенилаланина (одной из незаменимых аминокислот, входящих в состав белка).
При заболевании нарушаются обменные процессы, особенно важные для развивающегося мозга ребенка. В крови и других жидкостях организма накапливается в большом количестве фенилаланин и повышено образуются такие вещества как фенилпировиноградная, фенилмолочная и фенилуксусная кислоты, которые выделяются в повышенных количествах с мочой. Следствием нарушенного обмена в мозге является тяжелое психическое недоразвитие. Если не предпринято своевременное лечение, то больные на всю жизнь остаются глубокими инвалидами.
Поступающий в организм фенилаланин идет на построение белковой цепи или превращается в тирозин. Отсутствие в печени фермента фенилаланингидроксидазы препятствует нормальному превращению фенилаланина пищи в тирозин. Поэтому фенилаланин используется лишь при синтезе белка, а избыток накапливается в клетках печени и попадает в кровоток, где количество фенилаланина является токсичным для клеток мозга. Почки не справляются с его реабсорбцией, в результате чего он выводится с мочой. Именно наличие этого фенилкетона в моче дало основание назвать соответствующее патологическое состояние фенилкетонурией.
Заболевания пищевода
... микроскопические очаги кровоизлияний. Флегмонозный эзофагит развивается при инфицировании механических повреждений задней стенки пищевода и быстро распространяется по слизистой оболочке. Характеризуется появлением ... (некротический) эзофагит встречается при тяжёлом течении ряда инфекционных заболеваний (корь, скарлатина, сыпной тиф). Макроскопически обнаруживаются гнойно-кровянистые наложения ...
Варианты ФКУ
Фенилкетонурия 1.
Классическая фенилкетонурия (ФКУ) описана А.Folling.,1934г.
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена, локализующегося в длинном плече 12 хромосомы.
В основе болезни лежит дефицит фермента фенилаланин-4-гидроксилазы, обеспечивающего превращение фенилаланина в тирозин. В результате метаболического блока происходит значительное накопление в тканях и жидкостях больного организма фенилаланина и таких его производных, как фенилпировиноградная, фенилмолочная, фенилуксусная кислоты, фенилэтиламин, фенилацетилглютамин, и др.
В патогенезе ФКУ имеют значение следующие механизмы:
- Прямое токсическое действие на ЦНС фенилаланина и его производных;
- Нарушение в обмене белков, липо- и гликопротеидов;
- Расстройства транспорта аминокислот;
- Нарушение метаболизма гормонов;
- Нарушение обмена моноаминовых нейромедиаторов (катехоламинов и серотонина);
- Нарушение функции печени — диспротеинемия, генерализованная гипераминоацидемия, повышение ДФА, метаболический ацидоз, нарушение окислительной и белковосинтезирующей функции клеточных органелл.
Частота классической ФКУ среди новорожденных по данным массового скрининга в среднем колеблется от 1:5000 до 1:10000 по разным регионам России.
Фенилкетонурия 2.
Впервые атипичная ФКУ описана I.Smith, 1974г. Заболевание связано с дефицитом дигидроптеридинредуктазы.
Заболевание наследуется аутосомно-рецессивно. Генный дефект локализуется в коротком плече 4 хромосомы, участке 4р 15.3.
В результате недостаточности дигидроптеридинредуктазы нарушается восстановление активной формы тетрагидробиоптерина, участвующего в качестве кофактора в гидроксилировании фенилаланина, тирозина, и триптофана.
Частота заболевания составляет 1:100000 новорожденных.
Рано начатое лечение способствует нормализации фенилаланина в крови, однако не предупреждает появление клинической симптоматики, которая развивается в начале второго полугодия жизни. Фенилкетонурию 2 называют диеторезистентной ФКУ.
Фенилкетонурия 3.
Этот вариант болезни описал S. Kaufman в 1978 г. Заболевание наследуется аутосомно-рецессивно и связано с недостаточностью 6-пирувоилтетрагидроптерин синтетазы, участвующей в процессе синтеза тетрагидробиоптерина. Развивающиеся при этом расстройства сходны с нарушениями, наблюдаемыми при ФКУ 2.
Частота болезни составляет 1:30000 новорожденных. Фенилкетонурия 3 также диеторезистентна.
Другие варианты ФКУ:
Эти формы ФКУ связаны с нарушением альтернативных путей обмена фенилаланина. Формируется метилминдальная ацидурия и парагидроскифенилуксусная ацидурия.
- Материнская фенилкетонурия.
Заболевание развивается у потомков женщин, страдающих ФКУ и не получающих диету в зрелом возрасте. Патогенез мало изучен, предполагается, что он сходен с патогенезом остальных форм ФКУ. Тяжесть поражения плода коррелирует с уровнем фенилаланина в плазме матери. Так как эмбрион особенно чувствителен к тератогенным воздействиям , рекомендуется начинать диету еще до наступления беременности. В суточном рационе использовать менее 15-20 мг/кг фенилаланина. При это важно избегать дефицита незаменимых аминокислот.
Наследственные болезни среда и образ жизни человека
... генетически обусловленное врожденное нарушение обмена веществ, и что таким же образом могут развиваться и другие наследственные болезни. Дальнейшее понимание природы наследственных болезней связано с ... среды. Примером, показывающим насколько может варьировать проявление наследственной патологии человека, определяемой моногенно, является успешное лечение специальной диетой больных фенилкетонурией, ...
- Клиническая фенилкетонурия.
При рождении больные фенилкетонурией не отличаются от других новорожденных. Манифестация ФКУ происходит обычно в возрасте 2-6 месяцев.
Клинические проявления
Дети с фенилкетонурией (ФКУ) рождаются без каких-либо признаков болезни. Однако уже на втором месяце можно заметить некоторые физические признаки: посветление волос, радужек глаз, что особенно заметно у детей, родившихся с темными волосами. Многие дети очень быстро и чрезмерно прибавляют в весе, однако остаются рыхлыми, вялыми. У большинства из них рано зарастает большой родничек. Чаще всего явные признаки болезни обнаруживаются на 4-6 месяце жизни, когда дети перестают реагировать радостью на обращение к ним, перестают узнавать мать, не фиксируют взгляд и не реагируют на яркие игрушки, не переворачиваются на живот, не сидят. В течение многих лет соответствующим диагностическим тестом служит реакция между фенилпировиноградной кислотой, которая выделяется с мочой ребенка, и хлорным железом. При положительной реакции появляется типичное зеленое окрашивание. Кроме того, образуются и выводятся с мочой другие аномальные метаболиты, такие как фенилмолочная и фенилуксусная кислоты. Последнее соединение «пахнет мышами», так что болезнь легко диагностировать по запаху; именно так она и была впервые обнаружена.
По мере прогрессирования болезни могут наблюдаться эпилептиформные приступы — развернутые судорожные и бессудорожные типа кивков, поклонов, вздрагиваний, кратковременных отключений сознания. Гипертония отдельных групп мышц проявляется своеобразной «позой портного» (поджатые ноги и согнутые руки).
Могут наблюдаться гиперкинезы, атаксия, тремор рук, иногда парезы по центральному типу. Дети нередко белокурые со светлой кожей и голубыми глазами, у них часто отмечаются экзема, дерматиты. Обнаруживается склонность к артериальной гипотензии.
Первыми проявлениями болезни служат:
- вялость ребенка, отсутствие интереса к окружающему;
- повышенная раздражительность, беспокойство;
- срыгивание, рвота;
- судорожные эквиваленты: спонтанный рефлекс Моро, спонтанный рефлекс Бабинского, сосательные автоматизмы, приапизм у мальчиков, атетозные движения;
- судорожный синдром;
- заплесневелый, мышиный, волчий запах мочи и пота.
При отсутствии лечения формируется задержка статико-моторного и психоречевого развития, умственная отсталость достигает, как правило, глубокой степени (идиотия или имбецильность, глубокая психическая инвалидность).
Дигностика фенилкетонурии
Чрезвычайно важно установить диагноз в доклинической стадии или по крайней мере не позднее 2-го месяца жизни, когда могут проявиться первые признаки болезни. Для этого всех новорожденных обследуют по специальным профаммам скрининга, выявляющего повышение концентрации фенилаланина в крови уже в первые недели жизни. Оптимальные сроки обследования новорожденных — 5-14-день жизни . Каждого ребенка, у которого обнаруживаются признаки задержки развития или минимальная неврологическая симптоматика, необходимо обследовать на патологию обмена фенилаланина. Используют микробиологический и флюорометрический методы определения концентрации фенилаланина в крови, а также пробу Фелинга на фенилпировиноградную кислоту в моче (прибавление нескольких капель 5% раствора треххлористого железа и уксусной кислоты к моче больного приводит к появлению зеленой окраски пятна на пеленке).
Влияние развода на дальнейшую жизнь ребёнка
... смерти второго. Показатели уровня удовлетворённости жизнью последних были такими же, что и у "детей развода". Это наблюдение ещё раз подтверждает, что детям необходимы оба родителя. Вдвоем они ... в Швеции, где детству уделяется больше внимания со стороны государства, влияние разводов на последующую жизнь детей в некоторой степени нивелировано. Интересной представляется и прослеживаемая связь с ...
Эти и другие подобные методы относятся к категории ориентировочных, поэтому при положительных результатах требуется специальное обследование с использована ем точных количественных методов определения содержания фенилаланина в крови ц моче (хроматография аминокислот, использование аминоанализаторов и др.), которое осуществляется централизованными биохимическими лабораториями.
Дети требуют специального наблюдения и лечения в медико-генетических центрах (кабинетах поликлиник).
Дифференциальный диагноз проводят с внутричерепной родовой травмой, внутриутробными инфекциями.
ФКУ может быть диагностирована на основе обнаружения следующих признаков:
- o стойкой гиперфенилаланинемии (более 240 ммоль/л);
- o вторичного дефицита тирозина;
- o экскреции фенилкетонов с мочой (проба Феллинга на экскрецию фенилпировиноградной кислоты).
В настоящее время, согласно приказу Минздрава России № 316 от 30.12.93 проведение неонатального скрининга на ФКУ стало обязательным. Скрининирующие тесты должны быть простыми, недорогими и информативными.
Этим требованиям отвечают методы, используемые для ранней диагностики ФКУ:
- o микробиологический тест Гатри;
- o метод флюоресцирующих антител (лабораторный комплекс «Флюороскан», позволяющий проводить 800 проб в час);
- o метод тонкослойной хроматографии.
Оптимальные сроки обследования новорожденных — доношенных, зрелых — 5-6 день жизни; недоношенных, незрелых, больных — 10-14 день жизни.
Трактовка результатов:
1 группа.
Уровень фенилаланина не превышает 200 ммоль/л (1-3 мг%) — норма;
2 группа.
Уровень фенилаланина составляет 200-500 ммоль/л (3-10 мг%) — гиперфенилаланинемия. В эту группу входят дети с транзиторной гиперфенилаланинемией, вследствие незрелости ферментных систем печени и больные ФКУ. За данной группой проводится наблюдение по следующему плану: если в течение 6 недель при еженедельном исследовании уровень фенилаланина остается менее 500 ммоль/л, контроль за уровнем фенилаланина крови проводят до 1 года первоначально каждые 3 месяца, а затем каждые 6 мес. жизни. При уровне более 500 ммоль/л назначается диетотерапия.